医疗器械设计开发全流程解析:从概念到量产的合规之路
一件成功的医疗器械产品,从一个创新的概念到最终服务于患者,背后是一套严谨、复杂且高度规范化的设计与开发流程。这个过程不仅关乎技术的实现和功能的完善,更重要的是要确保产品在整个生命周期内都满足严格的法律法规要求。在中国,所有医疗器械的研发、生产和上市都必须严格遵循国务院颁布的《医疗器械监督管理条例》,这部核心法规为整个设计开发流程提供了根本的法律依据和框架。清晰地理解并执行这套流程,是企业控制风险、保证质量、顺利将产品推向市场的基石。
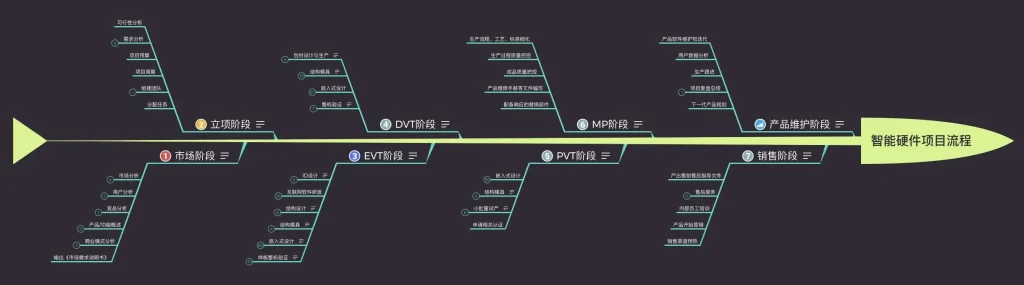
奠定基石:研发计划与可行性分析(ES阶段)
ES(Evaluation Specification)阶段是所有工作的起点,其核心任务是将一个初步的想法或市场需求,转化为一个清晰、可执行的研发项目。
项目提案与需求分析
项目的启动可能源于多种渠道:或许是临床一线医护人员提出的迫切需求,或是营销部门通过市场调研捕捉到的商业机会,也可能是公司为提升竞争力而制定的战略方向。无论来源如何,首要任务是进行深入的需求分析。这包括评估市场潜力、预估产品成本、并对标国内外的相关法规标准。
可行性评估与正式立项
在收集到足够的信息后,研发部门将牵头组织技术、质量、生产、法规等多方专家进行可行性评估。这个环节至关重要,它需要回答一个核心问题:我们的技术能力、资源配置和预估成本是否能支撑这个产品的成功开发?在这一初期阶段,就必须引入一个关键的合规步骤——医疗器械分类界定。明确产品属于第几类医疗器械,将直接决定后续法规路径的复杂程度、临床要求以及投入的资源。只有在可行性得到充分论证,且合规路径清晰后,项目才会被正式批准立项,并编制出纲领性的《产品开发计划书》。
从图纸到现实:工程验证与样品试制(EVT阶段)
EVT(Engineering Verification Test)阶段的目标,是将设计图纸转化为能够工作的实体原型,并验证设计的初步可行性。
详细设计与工艺开发
项目团队在接到开发任务后,会着手进行具体的产品设计,这包括结构、电路、软件等所有技术图样和设计文件的输出。与此同时,工艺设计也需同步进行,例如确定生产流程、设计专用工装夹具等。这两项工作并行,可以有效缩短开发周期。
样品制作与初步验证
此阶段会制作出少量的“工程样品”。这些样品的主要目的是验证产品的各项功能和性能是否符合设计初衷。通过一系列内部测试,如功能测试、寿命测试、安全测试等,开发团队可以识别并修正设计中存在的缺陷。这个过程的反馈是迭代改进产品设计的宝贵依据。值得注意的是,对于结构复杂或高风险的医疗器械,此阶段的验证结果也是后续进行医疗器械临床试验设计的重要参考。
精益求精:设计验证与小批量试产(DVT阶段)
DVT(Design Validation Test)阶段是承上启下的关键环节,其目的是验证经过初步修正后的设计,在模拟的生产环境下是否稳定、可靠,并为量产进行准备。
此阶段会进行小批量的试生产(例如200台)。与EVT阶段纯手工打造的样品不同,DVT阶段的产品会尽可能使用量产的模具和工装,在真实的生产线上进行组装。这不仅能进一步验证产品设计的成熟度,更能暴露出生产工艺、工装夹具、作业指导文件等方面可能存在的问题。质量部门会全面介入,对这批产品进行严格的检验,以确保其满足所有设计输入和法规要求。
量产前的大考:生产验证与量产准备(PVT阶段)
PVT(Production Verification Test)是进入大规模量产前的最后一道关卡,旨在验证生产线在全速运行状态下的稳定性和良品率。
在这一阶段,生产数量会进一步扩大(例如500台),完全模拟未来的量产状态进行生产。所有操作人员、设备、物料和生产环境都必须是正式量产的状态。此阶段的核心目标是验证整个生产体系的能力,确保整个质量管理体系和生产过程都符合《医疗器械生产质量管理规范》(即GMP)的严格要求,包括供应链的稳定性、生产效率、过程质量控制的有效性等。只有PVT阶段顺利通过,证明我们具备了稳定生产出合格产品的能力,才能为后续申请医疗器械生产许可提供有力的数据和文件支持,这也是产品获准上市销售的法定前提之一。
市场准入:正式量产与持续监督(MP阶段)
MP(Mass Production)意味着产品设计与开发阶段的结束,以及市场生命周期的开始。
技术资料转移与量产批准
在获得总经理的最终批准后,项目正式进入量产。研发部门会将全套最终版的设计图纸、工艺文件、检验标准等技术资料正式移交给生产、质量等相关部门。从此,产品的主要技术责任由研发端转移至生产端。
上市注册与持续改进
对于在中国市场销售的医疗器械,完成所有开发和验证工作,并整理形成完善的技术文档(即注册资料),是启动注册流程的基础。特别是对于风险较高的产品,完成第二、三类医疗器械注册并取得注册证,是产品合法上市销售的“身份证”。产品上市后,工作并未结束。企业还需要建立不良事件监测系统,持续收集市场反馈,并根据法规要求对产品进行必要的改进和迭代,履行贯穿产品全生命周期的监管责任。