病人监护产品(第二类)注册审查指导原则(2025年修订版)
 文档下载:病人监护产品(第二类)注册审查指导原则(2025年修订版)(2025年第27号)
文档下载:病人监护产品(第二类)注册审查指导原则(2025年修订版)(2025年第27号)
核心要点速览
2025年修订的《病人监护产品(第二类)注册审查指导原则》为多参数监护仪的注册申报提供了最新的技术指引。相比旧版,新原则在以下几个方面进行了重要的更新和明确:
- 明确适用范围与分类:适用于II类(07-04-01)病人监护仪,明确不包含III类及胎儿监护仪。
- 细化注册单元划分:预置式(一体机)和模块式(插件式)监护仪因结构差异大,必须划分为不同注册单元。
- 标准全面升级:强制要求符合GB 9706.1及YY 9706.249(多参数监护仪专用安全标准),同时需满足各参数模块(如血氧、血压)的专用标准。
- 强化临床准确度验证:对血压、血氧、体温等核心参数,明确要求提供临床准确度研究资料(遵循ISO 81060-2等标准)。
- 免临床评价路径:确认该产品属于免临床目录,重点在于提交与已上市同类产品的详细对比说明。
要点解读
病人监护仪是临床使用频率最高的医疗设备之一,集成了心电、呼吸、血压、血氧等多项生理参数监测功能。对于生产企业而言,病人监护产品注册是一项系统工程,涉及的标准繁多,技术要求复杂。
首先,在进行医疗器械分类和注册单元规划时,必须清晰界定产品的结构形式。指导原则明确指出,预置式和模块式监护仪应分开申报。这是因为模块式监护仪涉及模块插拔、通讯协议及扩展性,其风险点与一体机有显著不同。在第二、三类医疗器械注册策略制定中,合理的单元划分能有效避免测试和审评过程中的反复。
其次,医疗器械产品技术要求的编写需全面覆盖“1+N”的标准体系。“1”是指多参数监护仪专用标准YY 9706.249,“N”则是指各个监测模块对应的专用标准(如无创血压的YY 9706.230、脉搏血氧的YY 9706.261等)。特别注意的是,虽然产品本身免临床,但针对血压、血氧等参数,仍需提供“临床准确度”验证资料,这通常需要按照ISO 81060-2等标准进行小样本的临床数据采集或数据库验证。
最后,该产品属于免临床评价医疗器械目录范围。企业无需进行大规模确证性临床试验,只需提交同品种对比说明。对比的重点应放在各个监测参数的测量范围、精度、报警机制以及抗干扰能力上。若存在差异,需通过台架试验(如模拟器测试)证明差异不影响安全性与有效性。对于涉及网络连接和数据传输的监护仪,网络安全和软件研究资料也是审查的必查项。建议企业在申报前咨询专业的医疗器械注册咨询机构,协助梳理复杂的标准适用性和临床准确度验证方案。
指导原则原文
病人监护产品(第二类)注册审查指导原则(2025年修订版)
本指导原则旨在指导注册申请人对病人监护产品注册申报资料的准备及撰写,同时也为技术审评部门审查注册申报资料提供参考。
本指导原则是对病人监护产品的一般要求,注册申请人应依据产品的具体特性确定其中内容是否适用。若不适用,需具体阐述理由及相应的科学依据,并依据产品的具体特性对注册申报资料的内容进行充实和细化。
本指导原则是供注册申请人和技术审评人员使用的指导性文件,但不包括审评审批所涉及的行政事项,亦不作为法规强制执行,应在遵循相关法规的前提下使用本指导原则。如果有能够满足相关法规要求的其他方法,也可以采用,但是需要提供详细的研究资料和验证资料。
本指导原则是在现行法规和标准体系以及当前认知水平下制定,随着法规和标准的不断完善,以及科学技术的不断发展,相关内容也将适时进行调整。
一、适用范围
本指导原则适用于《医疗器械分类目录》中第二类病人监护产品,分类编码为07-04-01。本指导原则不适用于第三类病人监护产品。
本指导原则适用范围不包含母亲/胎儿多参数监护仪,但可参照本指导原则适用的相关条款执行。
二、注册审查要点
(一)监管信息
1.产品名称
产品的名称应符合《医疗器械分类目录》《医疗器械通用名称命名规则》《医疗器械通用名称命名指导原则》《医用诊察和监护器械通用名称命名指导原则》和相关法规、规范性文件的要求。例如:病人监护仪、病人监护系统、多参数监护仪等。
产品名称中不得存在型号规格描述,不得使用XX系列、XX型。
2.注册单元划分
注册单元划分应符合《医疗器械注册单元划分指导原则》的要求,原则上以产品的技术原理、结构组成、性能指标和适用范围为划分依据。
结构差异较大的产品,如预置式和模块式病人监护仪,应划分为不同的注册单元。
3.产品列表
以表格形式列出拟申报产品的型号、规格、结构及组成、附件,以及每个型号规格的标识(如型号或部件的编号,器械唯一标识等)和描述说明(如尺寸、材质等)。
(二)综述资料
1.概述
注册申请人应描述申报产品的通用名称及其确定依据,描述产品的管理类别、分类编码、适用范围。
如适用,申请人应描述有关申报产品的背景信息概述或特别细节,如:申报产品的历史概述、历次提交的信息,与其他经批准上市产品的关系等。
2.产品描述
2.1产品的结构和组成
申请人应描述产品的结构组成,提供产品主机、电源线、附件的图示和连接示意图,结合图示和连接示意图对产品的结构组成进行详尽描述。描述的内容包括主机和附件的结构、尺寸、与人体接触部分材料、接口、连接方式等。应结合产品控制面板和用户界面,对产品的功能(包括软件功能)、性能进行描述。
病人监护仪产品一般主要由主机和附件组成。附件包括电极、导联线、传感器,如:心电电极、心电导联电缆、血压袖带、血氧探头、体温探头、呼吸末二氧化碳气体测量组件等。
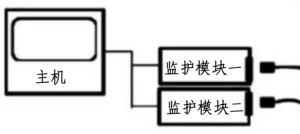
产品可按设计、型式、技术参数、附件及附加功能等不同分为若干型号,具有心电、无创血压、脉搏、血氧饱和度、体温、呼吸、呼吸气体(如:呼吸末二氧化碳)等监护单元,一般采用模块式和/或预置式结构。产品图示举例如下:
图1 预置式病人监护仪产品图示
图2 模块式病人监护仪产品图示
图3 预置式病人监护仪
图4 无创血压袖带
图5 血氧饱和度传感器
图6 体温探头
图7 模块式病人监护仪
图8 一次性使用心电电极2.2工作原理
病人监护仪产品通常包含多个不同生理监护单元,需分别描述不同生理监护单元的工作原理。
一般心电测量采用Ag/AgCl电极片放置在患者体表,通过获取患者心脏活动电位,记录各测量点间电位差得到心电信号。无创血压测量采用示波法,测出收缩压、平均压和舒张压、脉率值。呼吸测量采用胸阻抗法。呼气末二氧化碳浓度的测量则是利用CO2能吸收4.3μm波长的红外线原理,在呼出气体通路上,一侧用红外线照射,另一侧用传感器测出所接收红外线的衰减程度,依据红外光吸收率与二氧化碳浓度相关的原理测出CO2浓度。体温测量采用热敏电阻法或者通过探测器测量被测对象耳腔之间的红外辐射来显示被测对象的体温。脉搏血氧饱和度测量通常采用双波长脉动法。
2.3型号规格
对于存在多种型号规格的监护仪,应当明确各型号规格的区别。应当采用对比表或带有说明性文字的图片、图表,描述各种型号规格的结构组成(或配置)、功能、产品特征和运行模式、技术参数等内容。
2.4包装说明
应说明所有产品组成的包装信息。若附件为已注册或备案产品,应单独说明其包装信息。
若附件为无菌提供,应当说明其无菌屏障系统的信息;若附件具有微生物限度要求,应当说明保持其微生物限度的包装信息。说明如何确保最终使用者可清晰地辨识包装的完整性。
2.5研发历程
阐述申请注册产品的研发背景和目的。如有参考的同类产品或前代产品,应当提供同类产品或前代产品的信息,并说明选择其作为研发参考的原因。
2.6与同类和/或前代产品的参考和比较
列表比较说明申报产品与同类产品和/或前代产品在工作原理、结构组成、制造材料、性能指标、以及适用范围等方面的异同。
如适用,明确与申报产品联合使用实现预期用途的其他产品的详细信息。对于已获得批准的部件或配合使用的附件(如一次性使用心电电极、血氧传感器、体温探头、血压袖带等),应当提供注册证编号或备案号,以及国家药监局官方网站公布的注册或备案信息。
3.适用范围和禁忌证
3.1适用范围
申请人应根据产品适用人群、产品功能以及使用环境描述产品的适用范围。
适用范围举例:产品用于对成人、小儿和新生儿进行心电、呼吸、体温、脉搏血氧饱和度、无创血压、呼气末二氧化碳的监护,在医疗机构、转运中供培训合格的临床医护人员使用。
3.2预期使用环境
申请人应明确产品使用场所和使用环境要求。
产品预期使用场所包括:医疗机构、救护车等。
应明确可能影响其安全性和有效性的环境条件,如温度、湿度、压力、移动、振动、海拔等。
3.3适用人群
应根据临床验证资料明确产品适用的人群。如:适用于成人、小儿或新生儿。
3.4禁忌证
申请人应描述产品的禁忌证(包括绝对禁忌证、相对禁忌证),如不适宜使用的人群、疾病等情形。
4.产品的不良事件历史记录
注册申请人应关注并收集同类产品以及申报产品注册周期内的不良事件历史记录,可通过各国监管机构发布的不良事件资料库中查询相应不良事件数据。可以选择产品名称为方向开展医疗器械不良事件查询,通过在已选择的数据库的检索页面输入预期要进行查询的医疗器械产品名称,通过限制检索时间进行相关产品不良事件信息收集。
可以检索公开发布的不良事件信息如下:
4.1国家药品不良反应监测中心发布的“医疗器械不良事件信息通报”及“医疗器械警戒快讯”。
4.2美国不良事件查询数据库MAUDE、召回查询数据库Recall及按年份查询警告信(warning letter)。
4.3英国医疗器械警报(MHRA)。
4.4加拿大召回与警戒(Search recalls and safety alerts)。
4.5澳大利亚TGA不良事件(Database of Adverse Event Notifications – medical devices)、召回(System for Australian Recall Actions)及警戒(All alerts)。
4.6日本PMDA。
4.7德国BfArM。
(三)非临床资料
1.产品风险管理资料
申请人可参考GB/T 42062及YY/T 1437标准对产品进行全生命周期风险管理,提交风险管理资料。
参考GB/T 42062标准、适用的安全标准(如GB 9706.1、GB 9706.249等)及规范性文件,结合产品自身设计特点、临床用途及使用场景,充分识别与安全有关的特征,进行风险分析、评价及控制。风险控制的方案与实施、综合剩余风险的评价可参考GB/T 42062标准第7、第8章的相关要求。应确保各风险的可追溯性,提供各风险及综合剩余风险的可接受准则,确认各风险及综合剩余风险可接受。根据产品的可预见的事件序列识别危险情况和可能发生的危害,病人监护产品常见的事件序列示例见附表7-1。
2.医疗器械安全和性能基本原则清单
列明产品符合《医疗器械安全和性能基本原则清单》各项适用要求所采用的方法,以及证明其符合性的文件。对于《医疗器械安全和性能基本原则清单》中不适用的各项要求,应当逐项说明不适用的理由。
对于包含在产品注册申报资料中的文件,应当说明其在申报资料中的具体位置;对于未包含在产品注册申报资料中的文件,应当注明该证据文件名称及其在质量管理体系文件中的编号备查。
3.产品技术要求及检验报告
3.1申报产品适用标准情况
申报产品应当符合适用的强制性标准。若申报产品结构特征、预期用途、使用方式等与强制性标准的适用范围不一致,申请人应当提出不适用强制性标准的说明,并提供经验证的证明性资料。
3.2产品技术要求
产品技术要求的编制应符合《医疗器械产品技术要求编写指导原则》的要求。本条款给出产品需要满足的主要性能指标。注册申请人应参考相应的国家标准、行业标准,根据产品技术特点制定相应的性能指标要求。对于国家标准、行业标准中不适用条款,注册申请人应在研究资料的产品性能研究中说明理由。
3.2.1产品型号/规格及其划分的说明
应列明产品的型号、规格。对同一注册单元中存在多种型号、规格的产品,应明确不同型号、规格的划分说明。
病人监护产品通常包含软件组件,应明确软件的名称、型号规格(若适用)、发布版本、完整版本命名规则。软件模块(含医用中间件)若有单独的版本、版本命名规则均需说明。
3.2.2性能指标
产品的主要性能指标包括心电、呼吸、无创血压、脉搏血氧饱和度、体温、呼气末二氧化碳等生理参数的监护范围、精度等要求。
若病人监护产品中含有一次性使用心电电极、血氧传感器、体温探头等附件,应制定相应要求。应符合相应的产品标准和注册审查指导原则的要求,如YY/T 0196、GB/T 21417.1等。
软件功能:应符合《医疗器械软件注册审查指导原则(2022修订版)》的要求,明确软件的功能(核心功能(含安全功能)纲要)、使用限制、接口(如适用)、访问控制(如适用)、运行环境(如适用)、性能效率(如适用)。
安全要求:应符合GB 9706.1、YY 9706.249、YY 9706.108中规定的要求,或还应符合专用标准GB 9706.225、GB 9706.227、GB 9706.255、YY 9706.230、YY 9706.261、YY 9706.256中规定的要求。如适用于紧急医疗环境,还需符合YY 9706.112中规定的要求。
电磁兼容:应符合YY 9706.102及专用标准YY 9706.249中电磁兼容的要求,或还应符合专用标准GB 9706.225、GB 9706.227、YY 9706.230、YY 9706.261、YY 9706.256中关于电磁兼容的要求。如适用于紧急医疗环境,还需符合YY 9706.112中关于电磁兼容的要求。
3.3产品检验报告
注册申请人应提供产品检验报告,可以是医疗器械注册申请人出具的自检报告,也可以是委托有资质的医疗器械检验机构出具的检验报告。
检验报告应提供包含软件版本信息的软件用户界面图片,包括软件发布版本及软件完整版本。如无用户界面需列明软件完整版本。
如选择同一注册单元中的典型产品型号规格进行检验,应说明检验用型号规格的典型性。典型产品型号规格应是同一注册单元内能够代表本单元内其他型号规格产品安全性和有效性的产品型号规格。应考虑功能最齐全、结构最复杂、风险最高的型号。若一个型号规格不能覆盖其他型号规格的全部性能指标,可以选择多个典型型号规格,也可对其他型号规格不被能覆盖的差异部分进行检验。
4.研究资料
4.1性能研究
应当提供产品物理和/或机械性能指标的确定依据、设计输入来源以及临床意义,所采用的标准或方法、采用的原因及理论基础。对于适用的国家标准、行业标准中不适用的条款,说明不适用的理由。
4.1.1应说明产品的各项技术参数,包括监测参数、报警参数的调节范围及误差要求。
4.1.2应说明产品的报警功能,提交报警性能研究资料。
4.1.3若产品的生理参数监测功能应用了的新技术或其他的关键技术,应列明该技术的名称,说明其软件和/或硬件的实现方式,提供验证测试资料。
4.1.4若监护仪产品预期与其他医疗器械(如联合已注册或备案的附件使用)联合使用实现监护用途,应当提供证明联合使用安全有效的研究资料。
4.2电气系统安全性研究
应当提供电气安全、机械和环境保护以及电磁兼容性的研究资料,说明适用的标准以及开展的研究。
4.3软件研究
病人监护产品通常包含软件组件。申请人应按《医疗器械软件注册审查指导原则(2022年修订版)》的要求提供软件研究资料。
4.4网络安全研究
若申报产品具备电子数据交换、远程控制或用户访问功能,申请人应当按照《医疗器械网络安全注册审查指导原则(2022年修订版)》的要求,提供网络安全研究资料。
4.5生物学特性研究
病人监护产品具有多个应用部分,通常与人体皮肤接触,申请人应根据GB/T 16886.1系列标准对产品结构组成中与患者接触部分进行生物学评价。
若通过试验的方式开展评价,与人体完好皮肤表面短期接触的材料至少应考虑细胞毒性、致敏反应、皮内反应等评价终点。若包含与人体自然腔道接触的材料,应当考虑皮内反应是否能够涵盖对特定部分(口腔、直肠等)的刺激。
4.6清洁、消毒、灭菌研究
应明确主机和附件推荐的清洗和消毒工艺(方法和参数)、工艺的确定依据以及验证的相关研究资料。
若附件为无菌提供,应当明确灭菌工艺(方法和参数)和无菌保证水平(SAL),并提供灭菌验证及确认的相关研究资料。
若产品经消毒或灭菌后可能产生残留物质,应当对灭菌或消毒后的产品进行残留毒性的研究,明确残留物质信息及采取的处理方法,并提供相关研究资料。
若附件为使用者灭菌,应当明确推荐的灭菌工艺(方法和参数)、所推荐灭菌工艺的确定依据以及验证的相关研究资料。对可耐受两次或多次灭菌的产品,应当提供产品所推荐灭菌工艺耐受性的研究资料。
若附件为已注册或备案的产品,应明确按照附件说明书推荐的方法进行清洗、消毒或灭菌。
4.7稳定性研究
4.7.1货架有效期
若产品包含一次性使用附件,应当明确附件的货架有效期。应提供货架有效期和包装研究资料,证明在货架有效期内,在规定的运输贮存条件下,产品可保持性能功能满足使用要求。
4.7.2使用稳定性
应当明确主机的使用期限和附件的使用期限或使用次数。参照《有源医疗器械使用期限注册技术审查指导原则》提供产品使用稳定性的研究资料。可以对该产品进行使用状态列举,完整分析出临床使用的情况,直接进行产品的老化试验研究;也可以将产品分解为不同子部件(如主机、附件)进行评价。应详细分析分解关系,在此基础上采用适当的分解方式(如将产品分为关键部件及非关键部件等)确定产品的使用期限。
4.7.3运输稳定性
应当提供产品运输稳定性验证资料,证明在规定的运输条件下,运输过程中环境条件(例如:震动、振动、温度和湿度的波动)不会对医疗器械的特性和性能,包括完整性和清洁度,造成不利影响。如产品具有无菌提供的附件,应提供在宣称的有效期内以及运输条件下,产品包装保持完整性的验证资料。
应当提供产品环境试验研究资料,参考GB/T 14710等相关标准进行研究,评价产品在各种工作环境和模拟贮存、运输环境下的适应性,可以制定不同的气候环境条件和机械环境条件来进行试验,或通过对关键部件的试验来评价整机的情况,也可以通过已上市同类产品比对方式进行判断。也可以采用其他方法或者标准进行研究,但需说明理由。
4.8可用性研究
病人监护产品属于中、低使用风险医疗器械,应参考《医疗器械可用性工程注册审查指导原则》提交使用错误评估报告,包括基本信息、使用风险级别、核心要素、同类医疗器械上市后使用问题分析、使用风险管理、结论等内容。若前期已开展可用性工程工作,亦可提交可用性工程研究报告,用于替代使用错误评估报告。
4.9其他资料
4.9.1血压临床准确度评估
若产品具有无创血压监测功能,应当按《电子血压计(示波法)注册审查指导原则(2024年修订版)》要求提供血压临床准确度研究资料。
4.9.2血氧饱和度准确度评估
若产品具有脉搏血氧饱和度监测功能,应当按《脉搏血氧仪注册技术审查指导原则》要求提供脉搏血氧饱和度准确度研究资料。
4.9.3体温临床准确度评估
若产品具有临床体温测量功能(调整模式下运行的临床体温计),应当按《耳腔式医用红外体温计注册审查指导原则》、《医用红外额温计注册审查指导原则》相关要求提供体温临床准确度验证资料。
4.9.4光辐射安全研究
若产品包含光源(发出的光预期作用于人体),如血氧探头等,应当按《医疗器械光辐射安全注册审查指导原则》要求提供光辐射安全研究资料。
4.9.5免于临床评价资料
供患者的心电、无创血压、脉搏、血氧饱和度、体温、呼吸、呼吸气体监测用的病人监护产品列入《免于临床评价医疗器械目录》(以下简称《目录》)。应参照《列入免于临床评价医疗器械目录产品对比说明技术指导原则》提交申报产品与《目录》所述内容的对比资料及申报产品与《目录》中已获准境内注册医疗器械的对比说明。如经对比存在差异,应提交差异部分对安全有效性影响的分析研究资料。该资料应能证明申报产品与《目录》所述产品的基本等同性,若无法证明,应开展临床评价。
(四)产品说明书和标签样稿
产品说明书和标签应符合《医疗器械说明书和标签管理规定》和相关法规、规范性文件及相关标准的要求。还应关注说明书中的以下内容:
1.产品的性能指标应与产品技术要求中的一致;
2.应规定产品须由经过培训的专业人员操作;
3.应提示产品在同一时间仅限于一个患者使用;
4.应说明产品是否具备防除颤应用部分。若使用不具备防除颤效应的应用部分,应说明当除颤器用于患者时应采取的预防措施;
5.应说明产品是否可以和高频手术设备一起使用;
6.应规定电磁兼容方面相关的警告及措施;
7.应规定产品的清洁消毒方式及相关注意事项,说明维护保养的内容、周期及解决方法;
8.应公布产品常见故障和解决方法;
9.应说明关于报警限值的调整范围和报警阈值的设置建议。
(五)质量管理体系文件
应依据《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》的要求提供质量管理体系核查文件。
三、参考文献
- [1]中华人民共和国国务院.医疗器械监督管理条例:中华人民共和国国务院令第739号[Z].
- [2]国家市场监管总局.医疗器械注册与备案管理办法:国家市场监督管理总局令第47号[Z].
- [3]国家食品药品监督管理总局.医疗器械说明书和标签管理规定:国家食品药品监督管理总局令第6号[Z].
- [4]国家食品药品监督管理总局.医疗器械分类规则:国家食品药品监督管理总局令第15号[Z].
- [5]国家食品药品监督管理总局.医疗器械通用名称命名规则:国家食品药品监督管理总局令第19号[Z].
- [6]国家药品监督管理局.医疗器械通用名称命名指导原则:国家药监局通告2019年第99号[Z].
- [7]国家药品监督管理局.神经和心血管手术器械通用名称命名指导原则:国家药监局通告2021年第62号[Z].
- [8]国家食品药品监督管理总局.医疗器械分类目录:国家食品药品监督管理总局公告2017年第104号[Z].
- [9]国家食品药品监督管理局总局.医疗器械注册单元划分指导原则:国家食品药品监督管理总局通告2017年第187号[Z].
- [10]国家药品监督管理局.有源医疗器械使用期限注册技术审查指导原则:国家药监局通告2019年第23号[Z].
- [11]国家药品监督管理局医疗器械技术审评中心.医疗器械网络安全注册审查指导原则(2022年修订版):国家药品监督管理局医疗器械技术审评中心通告2022年第7号[Z].
- [12]国家药品监督管理局医疗器械技术审评中心.医疗器械软件注册审查指导原则(2022年修订版):国家药品监督管理局医疗器械技术审评中心通告2022年第9号[Z].
- [13]国家药品监督管理局.免于临床评价医疗器械目录:国家药品监督管理局通告2023年第33号[Z].
- [14]国家药品监督管理局.医疗器械临床评价技术指导原则:国家药品监督管理局通告2021年第73号[Z].
- [15]国家药品监督管理局.医疗器械产品技术要求编写指导原则:国家药品监督管理局通告2022年第8号[Z].
- [16]国家药品监督管理局.医疗器械注册申报资料要求和批准证明文件格式:国家药品监督管理局公告2021年第121号[Z].
- [17]国家药品监督管理局医疗器械技术审评中心.医疗器械安全和性能基本原则符合性技术指南:国家药品监督管理局通告2022年第29号[Z].
附表7-1
事件和情形示例
通用分类 事件和情形示例 要求 设计参数的不恰当规范:
可触及金属部分、外壳、应用部分、信号输入/输出部分等与带电部分隔离/保护不够,电介质强度不够,导致对电击危险防护不够,可能对使用者或患者造成电击危害;便携式提拎装置不牢固,带脚轮设备锁定不良,移动式设备易翻倒,设备支撑件强度不足,设备面、角、边粗糙,对飞溅物防护不够等可能对使用者或患者造成的机械损伤,血压袖带、血氧检测探头夹压力不合理引起的危险等;显示器辐射可能对操作者产生危害;对环境的电磁干扰超标,干扰其他设备正常工作;等等。
运行、性能要求不恰当规范:
各种参数正常监护范围设计的依据、各种参数报警设定值设计的依据、确保可靠报警采取的措施,等等。
与人体接触的部件:一次性心电电极、血氧探头、血压袖带等材料的生物安全性问题。
服务中的要求不恰当规范:
使用说明书未对设备及监护电极维护、保养方式、方法、频次进行说明,导致设备及各附件不能正常使用;等等。
寿命的结束:
使用说明书未对设备/附件的使用寿命和贮藏寿命进行规定,导致设备/附件超期非正常使用导致图像质量等性能指标降低,安全性能出现隐患;等等。制造过程 制造过程更改的控制不充分:
控制程序修改未经验证,导致设备性能参数指标不符合标准要求;等等。
制造过程的控制不充分:
生产过程关键工序控制点未进行监测,导致部件或整机不合格;等等。
供方的控制不充分:
外购、外协件供方选择不当,外购、外协件未进行有效进货检验,导致不合格外购、外协件投入生产;等等。运输和贮藏 不恰当的包装:
产品防护不当导致设备运输过程中损坏;等等。
不适当的环境条件:
在超出设备规定的贮藏环境(温度、湿度、压力)贮藏设备,导致设备不能正常工作;等等。环境因素 物理学的(如热、压力、时间):
过热环境可能导致设备不能正常工作;等等。
化学的(如腐蚀、降解、污染):
强酸强碱导致设备或监护电极损害;非预期使用于有麻醉剂的环境中,可能因为电气连接、设备结构、静电预防不良等引起混合气体爆炸;等等。
电磁场(如对电磁干扰的敏感度):
抗电磁干扰能力差,特定环境设备工作不正常;等等。
不适当的能量供应:
设备的供电电压不稳定,导致设备不能正常工作或损坏;等等。清洁、消毒和灭菌 未对消毒过程确认或确认程序不规范:
使用说明书中推荐的对与人体接触的附件的消毒方法未经确认,不能对监护电极进行有效消毒;等等。
消毒执行不恰当:
使用者未按要求对与人体接触的附件进行防护或消毒,导致院内感染;等等。处置和废弃 没提供信息或提供信息不充分:
未在使用说明书中对附件的处置和废弃方法进行说明,或信息不充分;未对设备废弃的处置进行提示性说明;等等。配方 生物相容性:
与人体接触的附件材料选择不当可致过敏等反应;等等。
与不正确配方有关的危害的警告不足;等等。可用性 设计缺陷引发可能的使用错误,如:
易混淆的或缺少使用说明书:
包括图示符号说明不规范、
操作使用方法不清楚、
技术说明不清楚、
未规定一次性使用电极等消耗性材料采购要求、
使用不适用的电极或传感器引起的危险、
清洁、消毒灭菌方法不明确、
重要的警告性说明或注意事项不明确等。
不适当的操作说明
副作用警告不充分:可用性 血压袖带、血氧检测探头夹压力不合理引起的危险,等等。
使用不当引起的风险:
由缺乏技术的/未经培训的人员使用,不能正确使用和维护保养设备;等等。
未按使用说明书规定使用指定监护电极;等等。
包括清洁消毒不当引起的危害、
使用未经消毒灭菌或不按规定的消毒灭菌方法进行消毒灭菌的器械、
一次性使用器械的多次使用、
不按制造商推荐的要求采购一次性使用的器械或传感器、
不能正常发挥使用性能等。
维护和校正不当,引起的不能正常发挥使用性能。功能性 由于老化、磨损和重复使用而致功能退化:
监护电极由于反复消毒、使用磨损等原因致老化、破损致监护电极带电;等等。附表7-2
相关产品标准
GB 9706.1 医用电气设备 第1部分:基本安全和基本性能的通用要求 GB 9706.225 医用电气设备 第2-25部分:心电图机的基本安全和基本性能专用要求 GB 9706.227 医用电气设备 第2-27部分:心电监护设备的基本安全和基本性能专用要求 GB 9706.255 医用电气设备 第2-55部分:呼吸气体监护仪设备的基本安全和基本性能专用要求 GB/T 14710 医用电器环境要求及试验方法 GB/T 16886.1 医疗器械生物学评价 第1部分:风险管理过程的评价与试验 GB/T 16886.5 医疗器械生物学评价 第5部分:体外细胞毒性试验 GB/T 16886.10 医疗器械生物学评价 第10部分:刺激与皮肤致敏试验 GB/T 16886.12 医疗器械生物学评价 第12部分:样品制备与参照样品 GB/T 21417.1 《医用红外体温计 第1部分:耳腔式》及第1号修改单 GB/T 25000.1 系统与软件工程 系统与软件质量要求和评价(SQuaRE)第1部分:SQuaRE指南 YY/T 0196 一次性使用心电电极 YY/T 0664 医疗器械软件 软件生存周期过程 YY 0828 心电监护仪电缆和导联线 YY 9706.102 医用电气设备 第1-2部分:基本安全和基本性能的通用要求 并列标准:电磁兼容 要求和试验 YY/T 9706.106 医用电气设备 第1-6部分:基本安全和基本性能的通用要求 并列标准:可用性 YY 9706.108 医用电气设备 第1-8部分:基本安全和基本性能的通用要求 并列标准:通用要求,医用电气设备和医用电气系统中报警系统的测试和指南 YY 9706.230 医用电气设备 第2-30部分:自动无创血压计基本安全和基本性能专用要求 YY 9706.249 医用电气设备 第2-49部分:多参数患者监护仪基本安全和基本性能专用要求 YY 9706.256 医用电气设备 第2-56部分:用于体温测量的临床体温计的基本安全和基本性能专用要求 YY 9706.261 医用电气设备 第2-61部分:脉搏血氧设备的基本安全和基本性能专用要求